



Resumen
Desde el brote de la enfermedad por coronavirus 2019 (COVID-19), los médicos han intentado todos los esfuerzos para comprender la enfermedad, y se ha identificado un breve retrato de sus características clínicas. En la práctica clínica, notamos que muchos pacientes con COVID-19 graves o críticamente enfermos desarrollaron manifestaciones clínicas típicas de shock, incluyendo extremidades frías y pulsos periféricos débiles, incluso en ausencia de hipotensión manifiesta. Comprender el mecanismo de la sepsis viral en COVID-19 es la garantía para explorar una mejor atención clínica para estos pacientes.
Con la evidencia recopilada de los estudios de autopsia sobre COVID-19 y la investigación científica básica sobre el coronavirus 2 del síndrome respiratorio agudo severo (SARS-CoV-2) y SARS-CoV, hemos presentado varias hipótesis sobre la patogénesis del SARS-CoV-2 después de múltiples rondas de debate entre investigadores de ciencias básicas, patólogos y clínicos que trabajan en COVID-19.
Presumimos que un proceso llamado sepsis viral es crucial para el mecanismo de la enfermedad de COVID-19. Aunque estas ideas podrían probarse imperfectas o incluso incorrectas más adelante, creemos que pueden proporcionar aportes y orientaciones para la investigación básica en este momento.
Introducción
El brote de coronavirus 2 del síndrome respiratorio agudo severo (SARS-CoV-2), que se informó por primera vez en Wuhan, China, en diciembre de 2019, ha tenido un enorme impacto en China y el mundo entero. La enfermedad causada por el SARS-CoV-2 se denomina enfermedad por coronavirus 2019 (COVID-19). Para el 19 de marzo de 2020, el número de casos confirmados había aumentado a más de 200 000. Aunque la mayoría de los pacientes infectados con SARS-CoV-2 tenían una enfermedad leve, aproximadamente el 5% de los pacientes tenían una lesión pulmonar grave o incluso disfunción multiorgánica, lo que resulta en una 1 · 4% de letalidad1.
En la práctica, notamos que muchos pacientes con COVID-19 graves o críticamente enfermos desarrollaron manifestaciones clínicas típicas de shock, incluyendo extremidades frías y pulsos periféricos débiles, incluso en ausencia de hipotensión manifiesta. Muchos de estos pacientes mostraron acidosis metabólica severa, lo que indica una posible disfunción de la microcirculación. Además, algunos pacientes tenían insuficiencia hepática2 y renal además de lesión pulmonar severa.
Estos pacientes cumplieron los criterios de diagnóstico para sepsis y shock séptico de acuerdo con el Sepsis-3 International Consensus 3 pero la infección por SARS-CoV-2 parecía ser la única causa en la mayoría de ellos.1 Los cultivos de muestras de sangre y del tracto respiratorio inferior resultaron ser negativos para bacterias y hongos en 76% de pacientes con sepsis en una cohorte COVID-19.
Por lo tanto, la sepsis viral sería más precisa para describir las manifestaciones clínicas de pacientes con COVID-19 graves o críticos.5 Comprender el mecanismo de la sepsis viral en COVID-19 es la garantía para explorar una mejor atención clínica para estos pacientes.
Infección por virus y patogenia de COVID-19 en órganos
En estudios de biopsia o autopsia, patología pulmonar para ambos principios6 y fase tardía7 los pacientes con COVID-19 mostraron daño alveolar difuso con la formación de membranas hialinas, células mononucleares y macrófagos infiltrando espacios aéreos, y un engrosamiento difuso de la pared alveolar. Se observaron partículas virales en las células epiteliales alveolares bronquiales tipo 2 mediante microscopía electrónica.
Además, la atrofia del bazo, necrosis de los ganglios linfáticos hiliares, hemorragia focal en el riñón, hígado agrandado con infiltración de células inflamatorias, edema y degeneración dispersa de las neuronas en el cerebro estaban presentes en algunos pacientes.
Se han aislado partículas de virus infecciosos del SARS-CoV-2 de muestras respiratorias, así como de muestras fecales y de orina (Zhao J, Guangzhou Medical University, comunicación personal) de pacientes con COVID-19, ¿lo que sugiere que la disfunción de múltiples órganos en COVID19 grave es causada? al menos parcialmente por un ataque directo del virus. Sin embargo, no hay informes sobre las observaciones post mortem de la amplia difusión de las partículas virales por autopsia en este momento.
Si el SARS-CoV-2 puede dirigirse directamente a otros órganos además del pulmón, especialmente aquellos órganos con alta expresión de la enzima convertidora de angiotensina 2 (ACE2) 12,13 y órganos con L-SIGN como posibles receptores celulares alternativos para el SARS-CoV-2, tiene que ser investigado más a fondo. Además, la cuestión de cómo se propaga el SARS-CoV-2 a los órganos extra-pulmonares sigue siendo un enigma. Se ha observado una variación genómica del SARS-CoV-2 circulante, y la diferencia en la virulencia necesita más investigación.
Respuesta inmune al SARS-CoV-2 y sepsis viral
Se ha demostrado que las citocinas y las quimiocinas proinflamatorias incluyen el factor de necrosis tumoral (TNF) α, interleucina 1β (IL-1β), IL-6, factor estimulante de colonias de granulocitos, proteína 10 inducida por interferón gamma, proteína quimioatrayente de monocitos,1 y las proteínas inflamatorias de macrófagos 1-α se elevaron significativamente en pacientes con COVID-19. Al igual que en una infección grave de influenza, la tormenta de citoquinas podría desempeñar un papel importante en la inmunopatología de COVID-19.
Estudios previos revelaron que las células epiteliales pulmonares, los macrófagos y las células dendríticas expresan todas las citocinas en cierta medida durante la infección por influenza a través de la activación de receptores de reconocimiento de patrones (incluidos los receptores Toll-like TLR3, TLR7 y TLR8), el gen I inducible por ácido retinoico y los miembros de la familia de receptores similares a NOD18.
Sin embargo, poco se sabe sobre la situación en COVID-19 en este momento. Es crucial identificar la fuente primaria de la tormenta de citoquinas en respuesta a la infección por SARS-CoV-2 y los mecanismos virológicos detrás de la tormenta de citoquinas. También sería relevante dilucidar la cinética de la activación de las citocinas durante la infección por SARS-CoV-2: ¿cuándo se liberaron las primeras citocinas y cuáles fueron?
Además, si el daño directo a los tejidos inducido por el virus, la tormenta sistémica de citoquinas o los efectos sinérgicos de ambos, contribuyen a la disfunción de múltiples órganos de los pacientes con COVID-19 grave, queda por abordar.
Además, vale la pena hacer un seguimiento acerca de si bloquear uno de estos mediadores proinflamatorios afectaría el resultado clínico. El anticuerpo monoclonal anti-IL-6R o los corticosteroides se han propuesto para aliviar la respuesta inflamatoria. Sin embargo, IL-6 podría desempeñar un papel importante en el inicio de una respuesta preliminar contra la infección por virus al promover la eliminación viral mediada por neutrófilos, ya que un estudio reveló que la deficiencia de IL-6 o IL-6R condujo a la persistencia de la infección por influenza y finalmente a la muerte en ratones.19 Y el uso de corticosteroides sigue siendo controvertido.
Sin embargo, la respuesta inmune desregulada también tiene una etapa de supresión inmune después de la fase proinflamatoria. Se caracteriza por una reducción sostenida y sustancial de los recuentos de linfocitos periféricos, principalmente células T CD4 y CD8 en pacientes con COVID-19, y se asocia con un alto riesgo de desarrollar una infección bacteriana secundaria. Esta condición, conocida como linfopenia, también se encontró en la influenza severa y otras infecciones virales respiratorias.
El mecanismo subyacente a la linfopenia sigue siendo desconocido. Estudios anteriores han demostrado que se detectaron partículas virales similares al SARS y el ARN del SARS-CoV en linfocitos T aislados de muestras de sangre periférica, bazo, ganglios linfáticos y tejido linfoide de varios órganos, lo que sugiere que el SARS-CoV podría infectar a las células T directamente. Los dominios de unión al receptor de las proteínas de pico entre el SARS-CoV-2 y el SARS-CoV muestran un alto grado de consistencia y también se detectó ARN del SARS-CoV-2 en muestras de sangre.
Por lo tanto, es razonable suponer que, además de la muerte celular inducida por la activación inducida por la interacción del ligando Fas y Fas, así como el eje del ligando inductor de apoptosis relacionado con el TNF.
El SARS-CoV-2 podría infectar directamente a los linfocitos, particularmente las células T, e iniciar o promo
ver la muerte celular de los linfocitos, lo que eventualmente conduce a linfopenia y respuestas antivirales dañadas. Sin embargo, tal hipótesis debe investigarse más a fondo.
También debe identificarse qué tipos de muerte celular están ocurriendo en los linfocitos después de la infección por SARS-CoV-2. Además, es intrigante que los linfocitos carezcan de expresión de ACE2, lo que sugiere un mecanismo alternativo por el cual el SARS-CoV-2 compromete a los linfocitos T.13 Si los macrófagos alveolares pueden o no fagocitar las partículas virales y luego transferirlas a los linfocitos es una pregunta abierta en el campo.
COVID-19 y coagulación anormal
Los estudios han revelado que el 71.4% de los no sobrevivientes de COVID-19 alcanzaron el grado de coagulación intravascular diseminada abierta (≥5 puntos según los criterios de la Sociedad Internacional de Trombosis y Hemostasia) y mostró resultados de coagulación anormales durante las etapas posteriores de la enfermedad; concentraciones particularmente aumentadas de dímero D y otros productos de degradación de fibrina se asociaron significativamente con un mal pronóstico4.
Sin embargo, los mecanismos concretos para la coagulopatía aún no están identificados. Si el SARS-CoV-2 es capaz de atacar directamente las células endoteliales vasculares que expresan altos niveles de ACE2,13
y luego conducen a una coagulación y sepsis anormales, aún debe explorarse.
Mientras tanto, ACE2 también es un importante regulador de la presión arterial. La alta expresión de ACE2 en el sistema circulatorio después de la infección por SARS-CoV-2 podría contribuir parcialmente a la hipotensión séptica.
Se han planteado preguntas sobre el uso de la terapia con inhibidores de los bloqueadores de los receptores de angiotensina II (BRA) e inhibidores de la ECA para pacientes con hipertensión arterial COVID-19. Algunos investigadores sugirieron que los inhibidores de la ECA podrían beneficiar a estos pacientes al reducir la inflamación pulmonar, aunque otros argumentaron que los inhibidores de la ECA podrían mejorar la entrada viral al regular los niveles de la ECA230. Sin embargo, ha habido poca evidencia clínica sobre el riesgo de tratar a los pacientes con COVID-19 con inhibidores de ARB o ACE. Se necesita más investigación para explorar si estos medicamentos inhiben o ayudan a la entrada viral.
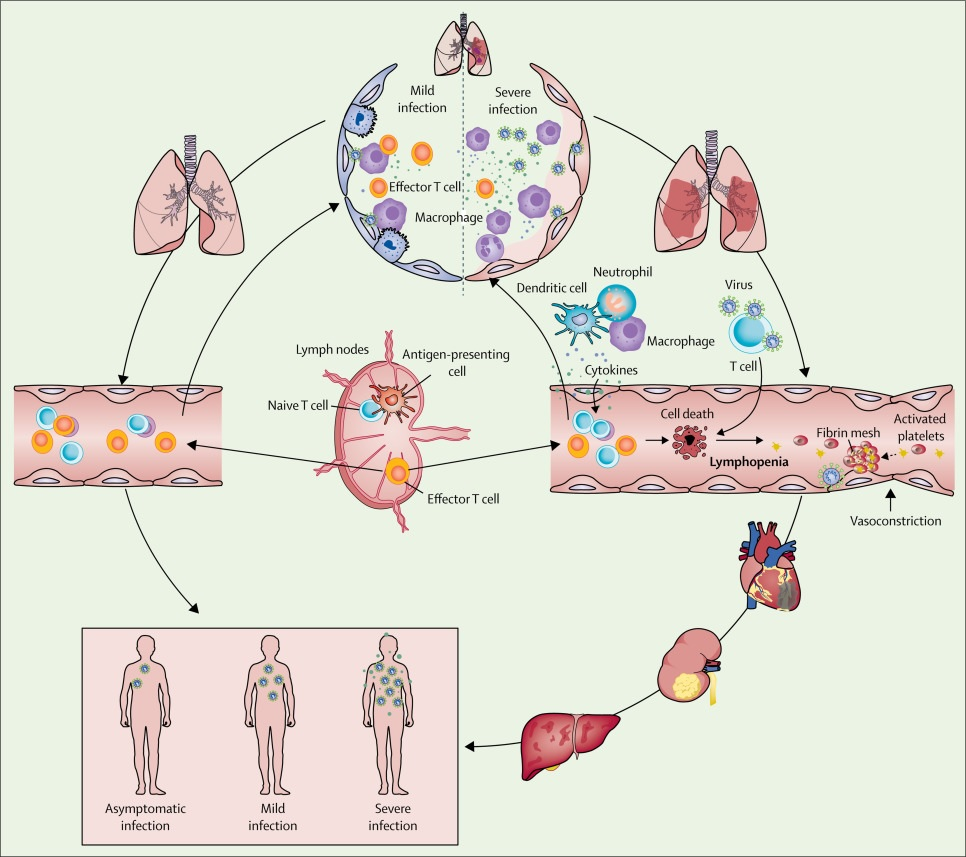
Figura: Ocurrencia y evolución del síndrome respiratorio agudo severo coronavirus 2 sepsis viral
Fuente: IntraMed


{kind=link}