



Se denomina miopatía inflamatoria idiopática (MII) a un conjunto de procesos que se caracterizan por una inflamación generalizada, no supurativa, del músculo estriado. Las MII son un grupo de enfermedades relativamente infrecuentes, con una incidencia anual de 0,8-8 casos/millón de habitantes/año. A nivel clínico, el hallazgo más frecuente es la debilidad muscular simétrica de predominio proximal. Las entidades más representativas del grupo de MII son la polimiositis (PM) y la dermatomiositis (DM) del adulto, la PM y DM de la infancia, la miositis asociada a otras enfermedades del colágeno, miositis asociada a neoplasias y la miopatía por cuerpos de inclusión. Recientemente se ha reconocido un subgrupo de DM en el que las manifestaciones cutáneas se presentan durante períodos prolongados en ausencia de enfermedad muscular, denominándose DM sine miositis o DM amiopática.
Aunque el principal órgano diana es el músculo, otros órganos internos se afectan con frecuencia, como la piel y el pulmón, por lo que las MII se consideran enfermedades sistémicas.
Pueden presentarse a cualquier edad, con dos picos de mayor incidencia: uno en la infancia (10-15 años) que corresponde a la DMS juvenil, y otro en la edad adulta (45- 60 años).
El tratamiento incluye la administración de glucocorticoides, inmunodepresores y puntualmente terapias biológicas.
Si bien la etiopatogenia no es bien conocida se ha avanzado en algunas teorías sobre la base de un agente externo físico, químico o infeccioso que actúa en un territorio genético predispuesto como la que se atribuyeron al influjo de la radiación UV como estímulo etiopatogénico. Otra teoría, es el fenómeno del microquimerismo fetal mediante el cual se cree que células inmunocompetentes del feto quedan anidadas en el seno materno y se activan produciendo una auténtica reacción de injerto contra huésped. Se han descrito casos de dermatomiositis en relación con prótesis de silicona en el contexto de un sustrato genético favorable. Se ha sugerido participación de diferentes fármacos (ej: estatinas), bacterias, parásitos, virus o determinantes genéticos en su patogenia. En ocasiones pueden asociarse a cáncer y la presencia de autoanticuerpos específicos y asociados a estas enfermedades sustenta la etiología autoinmune del proceso y ayuda a categorizar a los pacientes.
Se observa una alteración de la inmunidad tanto celular como humoral como lo prueba el acúmulo de linfocitos en el tejido muscular, la presencia de autoanticuerpos específicos dirigidos contra moléculas citoplasmáticas implicadas en la síntesis de proteínas y la respuesta a agentes inmunosupresores. Las citocinas que se producen en las células inflamatorias, en las células endoteliales o en las propias fibras musculares podrían ser responsables de la alteración de la función muscular. También se ha observado una sobreexpresión de moléculas HLA de clase I tanto en los miocitios regenerados como en las fibras no necróticas.
Por lo general, el compromiso cutáneo precede en unos 6 meses a la enfermedad muscular. No hay correlación entre el curso clínico de las lesiones cutáneas y la severidad de la miositis o de las manifestaciones sistémicas extramusculares de la DMS. Podemos distinguir lesiones patognomónicas como las Pápulas de Gottron, lesiones características como el Eritema en heliotro poque se observa hasta en el 60% de los pacientes con DM, lesiones más comunes en DM juvenil y lesiones raras en la DM. Otra lesiones caracteristicas son el Signo de Gottron.
En la Miositis por estatinas la presencia de anticuerpos anti-HMG-CoA reductasa sustenta el carácter autoinmune de esta patología. La expresión de HMG-CoA reductasa suele ser baja en la mayoría de los tejidos en circunstancias normales, pero estos se elevan marcadamente cuando las células musculares y de otros tipos son expuestas a las estatinas. Se ha sugerido que la sobreexpresión de HMG-CoA reductas asociada a la exposición a estatinas desencadena un proceso autoinmunitario en pacientes genéticamente susceptibles. A su vez, este proceso incrementa las células musculares en regeneración, produciendo niveles más altos de HMG-CoA reductasa y contribuyendo a que el proceso autoinmune se mantenga incluso después de descontinuar las estatinas. Por otro lado, el mecanismo de daño celulares un proceso poco entendido. La presencia del MAC en las membranas de células no necróticas sugiere un rol patogénico de los autoanticuerpos, lo cual también explica por qué los niveles de éstos se correlacionan con los niveles de CK y el grado de debilidad muscular.

El incremento de los niveles de las enzimas musculares y la presencia de autoanticuerpos son los datos más característicos de la DMS.
El daño muscular ocasiona un aumento de los niveles de enzimas musculares. La elevación de la creatinfosfokinasa (CPK) es el indicador más sensible y específico de la enfermedad muscular activa, presente en el 90% de pacientes. La aldolasa sérica es un indicador menos sensible para detectar la miositis activa, sin embargo, sus niveles pueden esta relevados en presencia de CPK normal. Las transaminasas hepáticas (GOT, GPT) y la lactatodeshidrogenasa (LDH) pueden estar elevadas, pero son poco específicas.
Los patrones de fluorescencia de los anticuerpos anti nucleres (ANA) obtenidos son tan solo sugestivos de los diferentes antígenos (ag), pero poco sensibles y no específicos. En caso de sospecha clínica de miositis, se debe proseguir el estudio por técnicas de inmunodetección específicas para ag. Un resultado positivo de ANA en los pacientes que presentan síntomas y signos sugestivos de tener una enfermedad inflamatoria muscular es solo una débil evidencia de PM/DM. Un resultado negativo tampoco descarta el diagnóstico de PM/DM.
La medición de los anticuerpos anti histidil-Trna sintetasa (Jo-1) sólo está indicada en los pacientes con ANA positivos con tinción citoplasmática y sospecha de PM/DM. Su positividad indica la existencia de una miositis inflamatoria idiopática, asociada a la presencia de otros anticuerpos anti-ARNt sintetasa hallados en pacientes con miositis que además presentan artritis, Fenómeno de Raynaud, fiebre y enfermedad pulmonar intersticial (EPI), y que definen un subtipo de patología conocido como Síndrome antisintetasa. Este síndrome ha venido siendo reconocido en los últimos años como una importante causa de miopatías inflamatorias. Usualmente la gravedad y la extensión de la enfermedad varían, siendo la miositis un poco menos severa en ausencia de este síndrome. Esta enfermedad es más frecuente en mujeres, con un promedio de edad de inicio a los 45 años. La morbimortalidad usualmente depende del compromiso pulmonar. El anticuerpo anti Jo-1 se asocia a un curso severo de la enfermedad y mal pronóstico. Aunque se ha descrito que los títulos pueden fluctuar e incluso desaparecer en las fases de remisión, no está indicada su medición para el seguimiento de la actividad de la enfermedad.
En términos generales podemos diferenciar entre anticuerpos específicos de miositisy autoanticuerpos asociados a miositis que pueden aparecer en otras colagenopatias y síndromes de superposición. Podemos encontrar autoanticuerpos dirigidos contra el RNA y ciertos antígenos citoplasmáticos relacionados con la síntesis de proteínas en el 60% de los pacientes con PM y DM. Este porcentaje es mayor cuando la miositis se asocia a otras colagenopatías.
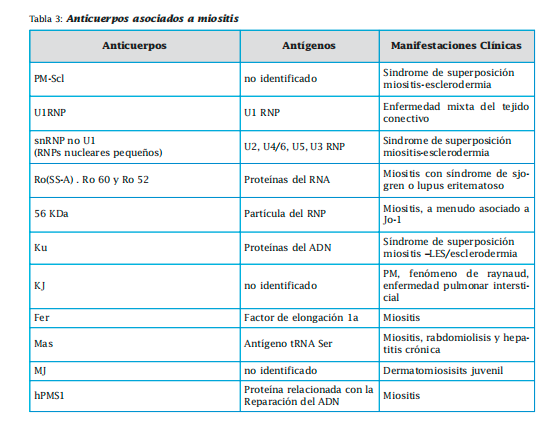
Entre los específicos se encuentran los anticuerpos antisintetasas (Jo-1, PL-7, PL-12, EJ, OJ, KS, Zo y Ha) y los no antisíntetasas. Dentro de estos últimos tenemos al SRP (partícula de reconocimiento de señal),anti-Mi2,anti-HMGRC,anti-MDAS, anti- 155/140, anti-140, anti-155 y anti SAE. Los anticuerpos asociados a miositis no son específicos de las miopatías. Los más importantes son antiPM-Scl, Ku, U1 RNP, RNP distintos al U1 ( U2-RNP, U4/U6-RNP y U5-RNP), Ro (SS-A), 56 KDa, KJ, Fer Mas ,MJ, hPMS1.
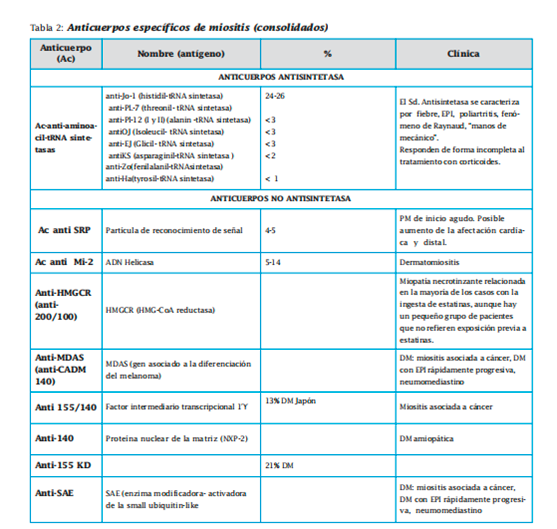
Sólo en un tercio de los pacientes se detectan anticuerpos específicos de miositis, siendo más frecuentes en aquellos casos no asociados a neoplasia. Estos anticuerpos están dirigidos contra el RNA de proteínas citoplasmáticas que intervienen en la síntesis proteica. Se ha observado que sus niveles se correlacionan con determinadas características clínicas, con la actividad de la enfermedad y tienden a desaparecer con su remisión.Podemos distinguir varios grupos, el más frecuente es el anti-Jo-1, presente en el 20% de DMS y en el 30-40% de PM y que se asocia con la presencia de síndrome antisintetasa. Los antiSRP se asocian con alteraciones cardíacas y enfermedad grave y los anti-Mi 2 son propios de la DMS (signo de la “V” del escote, signo de chal, engrosamiento de las cutículas, artralgias, artritis, fenómeno de Raynaud, EPI y ausencia de enfermedad cardíaca). Recientemente se ha descrito la presencia de Ac anti-155 Kd en el 21% de pacientes con DM. Su presencia se asocia a un factor genético de riesgo HLA DQA1*0301 a un aumento de la frecuencia del rash en V, y los pacientes presentan manifestaciones clínicas diferentes de aquellos con anticuerpos antisintetasa. Los pacientes con anti- 155/140 representan el 13% de pacientes con DMS en la población japonesa. Parecen ser muy específicos de esta enfermedad, y se asocian a eritema flagelado y a neoplasia, además del signo de Gottron y eritema en heliotropo. También se ha descrito la asociación del Ac anti-CADM-140 como marcador de DM amiopática, en un subgrupo de pacientes caracterizado por baja asociación de afectación muscular y de neoplasia subyacente, pero con mayor riesgo de vasculopatía y de EPI rápidamente progresiva.
En la evaluación inicial de todos los pacientes con sospecha de MI debe incluirse:
• Laboratorio:
– Determinación de enzimas procedentes del músculo como la CPK, ALT (GPT), AST (GOT), LDH y aldolasa. Otras determinaciones que pueden ser útiles para valorar el estado general del paciente y además descartar otras causas de miopatía incluirían: glucemia, creatinina, urea, Na, P, Ca, P, hemograma, VSG, PCR, TSH y sedimento urinario.
– Estudio de autoanticuerpos:Debe incluir la determinación de ANA, anti-DNA, anti-Sm, anti-RNP, anti-Ro/La, anti-cardiolipina y anticoagulante lúpico. De los anticuerpos específicos el anti-Jo-1. También es útil la determinación de anti PM-Scl para valorar la asociación a esclerodermia. La determinación de otros autoanticuerpos dependerá de las manifestaciones clínicas y de la disponibilidad de un laboratorio que pueda realizarlos.
– Medición de los niveles de complemento .
– Serología infecciosa: HIV, VHC, Trichinella spiralis, Toxoplasma, Borrelia burgdorferi, Parvo-virus B 19.
– Marcadores tumorales (su determinación sistemática es discutible): CA-125, CA 19.9, CA 15,3, alfa-fetoproteína, antígeno carcino embrionario, PSA.
• Rx de tórax PA y lateral.
• Pruebas de función respiratoria y DLCO.
• Saturación de oxígeno por pulsioximetría.
• Electrocardiograma.
• Ecografía abdominal.
• Electromiograma.
• Biopsia muscular.
• Radiografías de articulaciones afectadas y de zonas con calcinosis.
• Exploración ginecológica y ecografía pélvica (descartar neoplasia ovárica).
• Exploración mamaria y mamografía.
• Exploración ORL.

El diagnóstico diferencial es amplio e incluye las enfermedades musculares hereditarias, los trastornos del tiroides, que no sólo se pueden confundir con la polimiositis, sino que también pueden aparecer y coexistir con las miopatías inflamatorias condicionando una mala evolución y una respuesta terapéutica inadecuada; las infecciones parasitarias, como la toxoplasmosis o la triquinosis, y el consumo de fármacos, en especial los fármacos utilizados en el tratamiento de las dislipemias, como las estatinas o los fibratos, pero también los glucocorticoides o los antipalúdicos de síntesis utilizados en el tratamiento de algunas manifestaciones de la enfermedad. Otro aspecto a tener en cuenta es el diagnóstico erróneo de hepatitis por elevación de las transaminasas, ya que estas enzimas también pueden ser de origen muscular y se elevan junto a la creatinquinasa en las miopatías. Entre las distrofias, el déficit de disferlina, proteína de membrana de la fibra muscular y que participa en la reparación celular, es lo que con mayor frecuencia puede confundirse con una polimiositis. La sospecha y la confirmación diagnóstica de esta entidad son importantes, ya que parece que el tratamiento con glucocorticoides tiene un efecto negativo en el músculo, que empeora el cuadro clínico y la evolución.
Autora: Dra. Patricia Gentili – Jefa Área Inmunología – Fares Taie Instituto de Análisis
Contacto: inmunología@farestaie.com.ar


{kind=link}